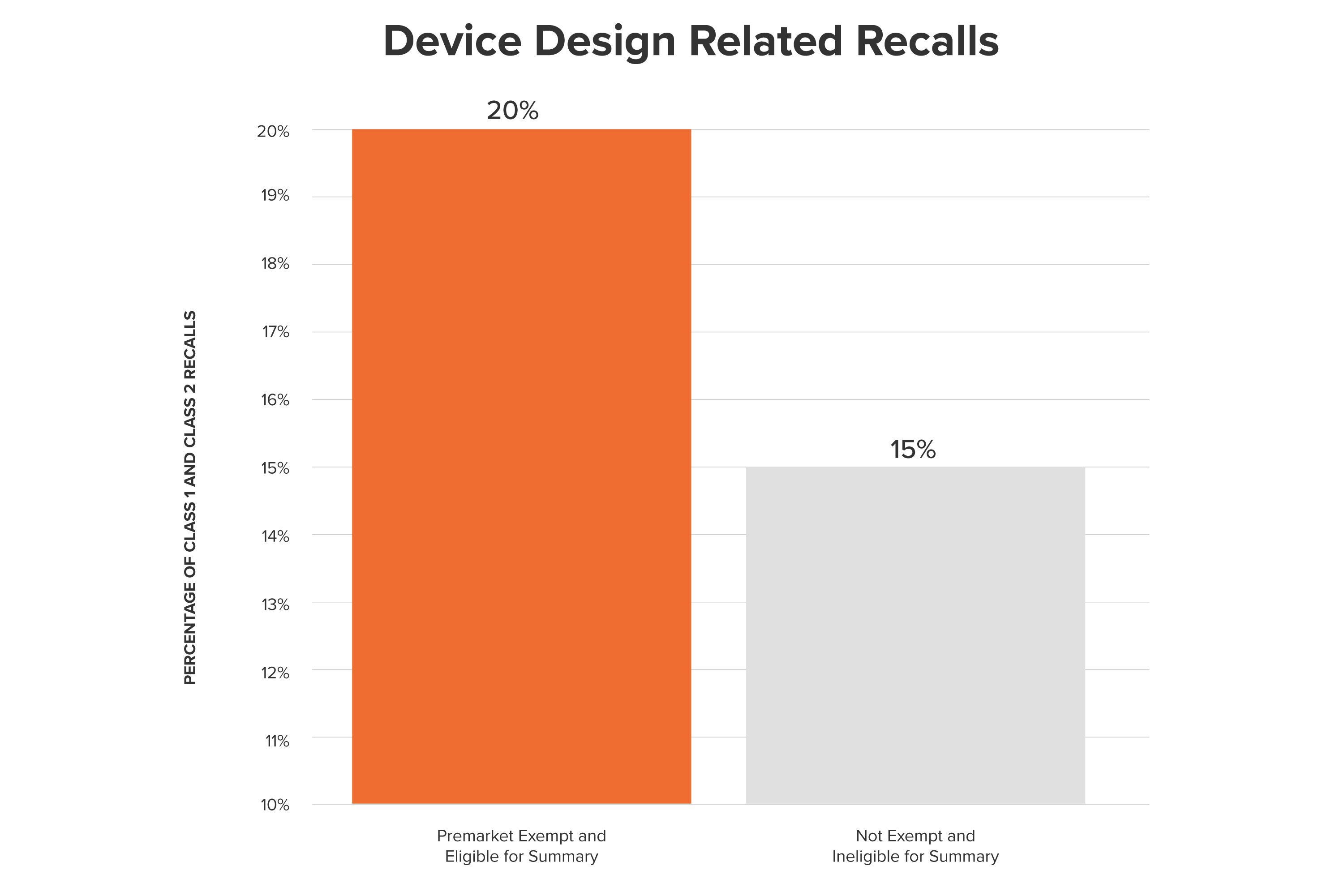

A core part of the FDA’s job requires balancing the tension between streamlining medical product innovation, and patient safety.
To encourage faster innovation, and in response to the 21st Century Cures Act, the FDA recently revised the 510(k) application process to reduce regulatory burden. Now more Class II (described by the FDA as presenting “a moderate risk of harm”) medical devices can bypass the 510(k) regulatory process and go directly to market. They join over 2,100 Class I (“minimal potential for harm”) and Class II product classes previously added to the Premarket Exemption list.

